Article Text

Abstract
Among other key objectives, the 2009 Family Smoking Prevention and Tobacco Control Act was designed to end an era of constant product manipulation by the tobacco industry that had led to more addictive and attractive products. The law requires new tobacco products to undergo premarket review by the US Food and Drug Administration (FDA) before they can be sold. To assess FDA's implementation of its premarket review authorities, we reviewed FDA actions on new product applications, publicly available data on industry applications to market new products, and related FDA guidance documents and public statements. We conclude that FDA has not implemented the premarket review process in a manner that prioritises the protection of public health. In particular, FDA has (1) prioritised the review of premarket applications that allow for the introduction of new tobacco products over the review of potentially non-compliant products that are already on the market; (2) misallocated resources by accommodating the industry's repeated submissions of deficient premarket applications and (3) weakened the premarket review process by allowing the tobacco industry to market new and modified products that have not completed the required review process.
- Public policy
- Advocacy
- Public opinion
- Surveillance and monitoring
- Tobacco industry
Statistics from Altmetric.com
In 2009, the US Congress passed the Family Smoking Prevention and Tobacco Control Act (Tobacco Control Act), providing the US Food and Drug Administration (FDA) with the authority to regulate tobacco products. As part of that authority, Congress provided that no new regulated tobacco products could enter the market without first undergoing review by FDA. In a compromise negotiated with the tobacco industry, the law ‘grandfathers’ tobacco products that were already on the market.1 Products that were commercially available at the time the law was introduced, and have not been changed in any meaningful way, do not require FDA authorisation to stay on the market. However, the law mandates that a manufacturer submit to FDA review before any new product, including new versions of previously available products, can be sold at retail.2 One aim of the requirement is to address the tobacco industry's history of manipulating its products to maximise addictiveness and increase attractiveness to consumers, and to prevent more harmful products from ever entering the market.3–8
In the almost 7 years since the enactment of the Tobacco Control Act, FDA has failed to implement the premarket review process in a manner that maximises the protection of public health. Instead, as explained in this Special Communication, the agency has misplaced its priorities, and thereby has undermined the potential public health benefits of tobacco regulation, in three distinct ways. First, rather than prioritise the removal of non-compliant products from the marketplace, FDA has given precedence to the review of applications that allow for the introduction of new tobacco products. Second, FDA has accommodated the tobacco industry's repeated submission of deficient premarket applications, rather than dismissing such flawed applications outright or allowing only reasonable amendments. Finally, even though industry marketing activities are widely publicised, FDA has failed to prioritise the enforcement of premarket review against companies that have avoided the process entirely and introduced new or modified products to the market without authorisation. These conclusions are based on our review of FDA actions on new product applications, publicly available data on industry applications to market new products, and the agency's guidance documents and public statements.
Background on the Tobacco Control Act's premarket review provisions
The cut-off date for products that are grandfathered and do not require FDA review is 15 February 2007.1 Any new or modified product introduced after that date must be authorised by FDA before it can be sold. This includes any entirely new brand or sub-brand of a product, as well as any modification to a legally marketed product.1 Whether FDA will authorise a new product to be sold depends on the manufacturer's ability to demonstrate that it has satisfied the criteria for one of the regulatory pathways for new products (figure 1). Under the Premarket Tobacco Product Application (PMTA) pathway, the manufacturer must show that introduction of a new product would be ‘appropriate for the protection of the public health’, taking into account ‘the risks and benefits to the population as a whole, including users and nonusers of the tobacco product’.9 In essence, this requires the applicant to show that, on balance, allowing the sale of the new product would likely reduce tobacco-related harms. The Substantial Equivalence (SE) pathway provides for less rigorous review if a manufacturer can show that its product is nearly the same as a predicate grandfathered product. When this pathway is being used, FDA's task is to determine whether the product is different from the predicate in any way that raises ‘different questions of public health’.10 If so, the SE pathway is not available, and the product must go through PMTA review before it can be sold. By contrast, if the new product does not raise any ‘different question of public health’, FDA will issue an SE order permitting that product to be marketed.

Premarket review pathways. FDA, US Food and Drug Administration; SE, Substantial Equivalence; Premarket Tobacco Product Application.
Although the general rule is that premarket review is required for both the PMTA and SE pathways, the Tobacco Control Act created an exception for SE applications (also termed ‘SE Reports’ by FDA) submitted by 22 March 2011.2 ,11–13 Products with SE applications submitted by that date are permitted to be sold while their applications are under review. FDA calls these applications ‘Provisional’ SE applications, because the products are provisionally on the market.11–17 The industry is free to manufacture and sell these products, but if FDA later determines that such a product is not substantially equivalent to a predicate product, it must be removed from the market.
While FDA's SE review process mimics the agency's SE review of modified medical devices, the administration of that authority ought to look very different in the tobacco context. On the medical device side, FDA's mission overlaps significantly with the regulated industry's goal: to facilitate the provision of safe and effective devices to consumers. It is in the public's interest to get modified medical devices to market as quickly as possible (so long as they are safe and effective), and it is in a device maker's best interest to collaborate with FDA to ensure that its devices have minimal to no unintended consequences. The bad publicity and potential legal liability that comes from a device that causes more harm than good can be exceptionally damaging to a device maker.18
By contrast, the SE review process for tobacco products is not designed to ensure that modified products are safe; it is instead intended to ensure that any changes to tobacco products do not create additional public health harms. However because tobacco products are already so harmful, the tobacco industry has little incentive to ensure that this review is effective. The industry will not be penalised by the marketplace for selling products that are marginally more harmful, and because its products are addictive, the industry's only incentive is to maximise product availability, addictiveness and appeal, and thereby increase sales. Furthermore, unlike in the medical device context, there is no public benefit to rushing new tobacco products to market. Public health gains are likely to accrue only if the SE requirements are rigorously applied and the industry is forced to go through the more rigorous PMTA process when appropriate. Thus, FDA and the tobacco industry do not share a common set of interests; FDA's goal should be to ensure that the SE requirements are scrupulously enforced, while the industry has every incentive to evade those same requirements. Yet, FDA's implementation of the SE review process for tobacco has reflected priorities that are more suitable for the review of devices, where it is an appropriate goal to move new or modified products to the market as quickly as possible. These misplaced priorities are having a significant negative impact on public health.
Misplaced priority #1: FDA prioritises the introduction of new tobacco products over the removal of non-compliant ones
The tobacco industry submitted 3517 Provisional SE applications, nearly all of them in the final few weeks before the 22 March 2011 deadline.17 ,19 This avalanche of applications dwarfs the number of submissions to all other new product pathways in the 5 years since that deadline, including ‘Regular’ SE applications (those submitted after 22 March 2011) (figure 2). FDA has focused its review on Regular SE applications rather than Provisional SE applications. This prioritisation has important implications for public health because allowing new products onto the market through the Regular SE pathway will have no beneficial impact on public health. However, the removal of Provisional SE products from the market could secure public health gains, as any non-compliant products would be found to have raised different questions of public health, and thus, have the potential to pose new harms to public health. Prioritising Regular SE applications serves the tobacco industry's interest in getting new products to the market as quickly as possible, and further delays agency action on those products that are already on the market. Given FDA's mandate to protect public health in its regulation of tobacco products, FDA should prioritise the review of Provisional SE applications instead.

Premarket submissions to FDA (data source: U.S. Food and Drug Administration, http://www.accessdata.fda.gov/FDATrack/track?program=ctp&id=CTP-OS-total-productsubmissionsreceived&fy=all). FDA, US Food and Drug Administration; PMTA, Premarket Tobacco Product Application; SE, Substantial Equivalence.
FDA's prioritisation of Regular SE applications over Provisional SE applications can be confirmed by examining FDA's public data regarding the status of its reviews. The multistep SE review process concludes with a scientific review, analysing the differential characteristics of the new and predicate products.12 ,14–16 ,20–22 The number of applications reaching this final stage provides a clear picture of FDA's priorities.17 ,23 Of the 3517 Provisional SE applications that have been submitted, FDA has only initiated scientific review of 645 of them, about 18%.17 By contrast, it has initiated scientific review of 1904 of the 1917 Regular SE applications that it has received, more than 99%.23 Moreover, of the 645 Provisional SE applications that are currently under review, FDA initiated the review of the vast majority (604) in 2014 or 2015.17 While this progress is a positive development, all these applications were submitted in March 2011. For Regular SE applications, scientific review on the first applications began in May 2012, and the agency has steadily initiated review of new applications since then.23
FDA has also publicly confirmed that it is prioritising Regular SE applications, explaining that this focus is driven by the fact that Regular SE applications represent products that are not currently on the market.12 ,14–17 ,19 ,23 There is no public health justification for trying to move new products to the market as quickly as possible. There is no statutory deadline by which FDA must review Regular SE applications, and focusing on the introduction of new products rather than the removal of non-compliant ones is inconsistent with the goals of the Act, and does not protect public health.
The pace of FDA review further exacerbates this problem. If the agency were acting quickly on Regular SE applications, it could eventually clear the backlog of Provisional SE applications, despite not prioritising them. Although the FDA has taken action to speed up its review, steady action on Regular SE applications and action on a handful of Provisional SE applications has not yet significantly reduced the backlog, with the number of SE applications pending before the agency having remained above 3,500 since March 2011 (figure 3).19 FDA has established performance measures for its review of Regular SE applications and plans to reach a point whereby it would begin its review of all new SE applications as soon as they were submitted.24 Notably, however, the agency has not established any performance measures for its review of Provisional SE applications, claiming that it lacks sufficient experience with them.12 ,25 This justification is non-sensical because the only differences between Provisional and Regular SE applications are (1) the date that the agency received them and (2) the fact that Provisional SE products are already on the market. Because of the agency's slow pace of review and focus on Regular SE, rather than Provisional SE applications, FDA has permitted unauthorised products to remain on the market. Indeed, for more than 5 years, as many as 3000 tobacco products have been in stores, gaining a foothold in the market, without any determination by FDA that they have met the proper legal standards.

Status of premarket submissions to FDA (data source: U.S. Food and Drug Administration, http://www.accessdata.fda.gov/FDATrack/track?program=ctp&id=CTP-OS-total-productsubmissionsreceived&fy=all, http://www.fda.gov/TobaccoProducts/Labeling/MarketingandAdvertising/ucm339928.htm). FDA, US Food and Drug Administration; SE, Substantial Equivalence.
Reviewing Provisional SE applications might not need to be a top priority if the tobacco industry could be trusted to submit applications only for products that were, in fact, nearly identical to the grandfathered predicate products. Given the tobacco industry's history of deception, however, this is not an assumption that can safely be made. Moreover, the substantial number of SE submissions received by FDA immediately before the March 2011 deadline suggests that instead of making a good-faith effort to determine which products were ‘substantially equivalent’ to grandfathered products, the tobacco industry instead sought to keep as many products on the market as possible, regardless of a product's eligibility for the SE pathway. Indeed, until September 2015, every final FDA decision on a Provisional SE application had found that the product at issue was not substantially equivalent (NSE) to the predicate product, as the application had claimed (figure 3). In September 2015, FDA did grant SE orders to 98 products that had undergone a new streamlined review process for Provisional SE applications with only labelling or packaging changes.i 26 However, the remaining backlog of more than 3000 Provisional SE applications should prompt alarm that there are likely more Provisional SE products still on the market that do not meet the statutory requirements. Use of these potentially non-compliant products is likely increasing overall health harms from tobacco use.
Misplaced priority #2: FDA is squandering resources by repeatedly accommodating the industry's submission of deficient premarket applications
It is unclear to what extent the slow pace of FDA's review of SE applications is due, in part, to unnecessary and unwarranted accommodation of the tobacco industry. Under the Tobacco Control Act, it is the applicant's responsibility to demonstrate compliance with the prerequisites for a marketing order under either PMTA or SE pathway. Although FDA can and should assist applicants with understanding the requirements of the Tobacco Control Act, FDA has squandered resources and further delayed the removal of illegitimate tobacco products from the market by making numerous accommodations to tobacco companies that submitted clearly deficient premarket applications.
This excessive level of accommodation is illustrated by the agency's first action on Provisional SE applications, which was taken in February 2014. The four applications at issue, submitted by the same manufacturer, failed to identify a predicate product to serve as the comparison for the new products.27 Because an SE application focuses on the differential characteristics of the predicate and new product,10 the failure to identify a predicate product should represent a fatal deficiency in an SE application. However, FDA did not contact the manufacturer to request the identification of the missing predicate products until 19 March 2013, roughly 2 years after the applications were submitted.27 The agency had eight unsuccessful follow-up communications, with the applicant unable to adequately identify the predicate.27 FDA finally issued NSE orders on 20 February 2014, removing the products from the market almost 3 years after the agency received the applications.27
FDA's extended dialogue regarding a fatally deficient application reveals an important issue. At least some of the manufacturers submitting SE applications either have a significant misunderstanding of the most basic requirements of the process, or they are deliberately attempting to exploit the process. While it is possible that manufacturers did not understand the SE process by the provisional deadline, that is unlikely given the clear guidance provided by FDA regarding what must be included in an SE application.28 It seems more probable that some manufacturers acted in bad faith and knowingly submitted deficient applications. It would be reasonable for the agency to allow an amendment or two to the scientific information included in an application. But as the agency's information shows, many of the deficiencies focus on the mere identity of the predicate or new product, which ought not be pieces of information that require multiple follow-ups, let alone eight or more, as has often been the case. In any event, whether the manufacturer was ill-informed or flouting the law, it is FDA's responsibility to quickly reject fatally flawed applications and remove non-compliant products from the market.
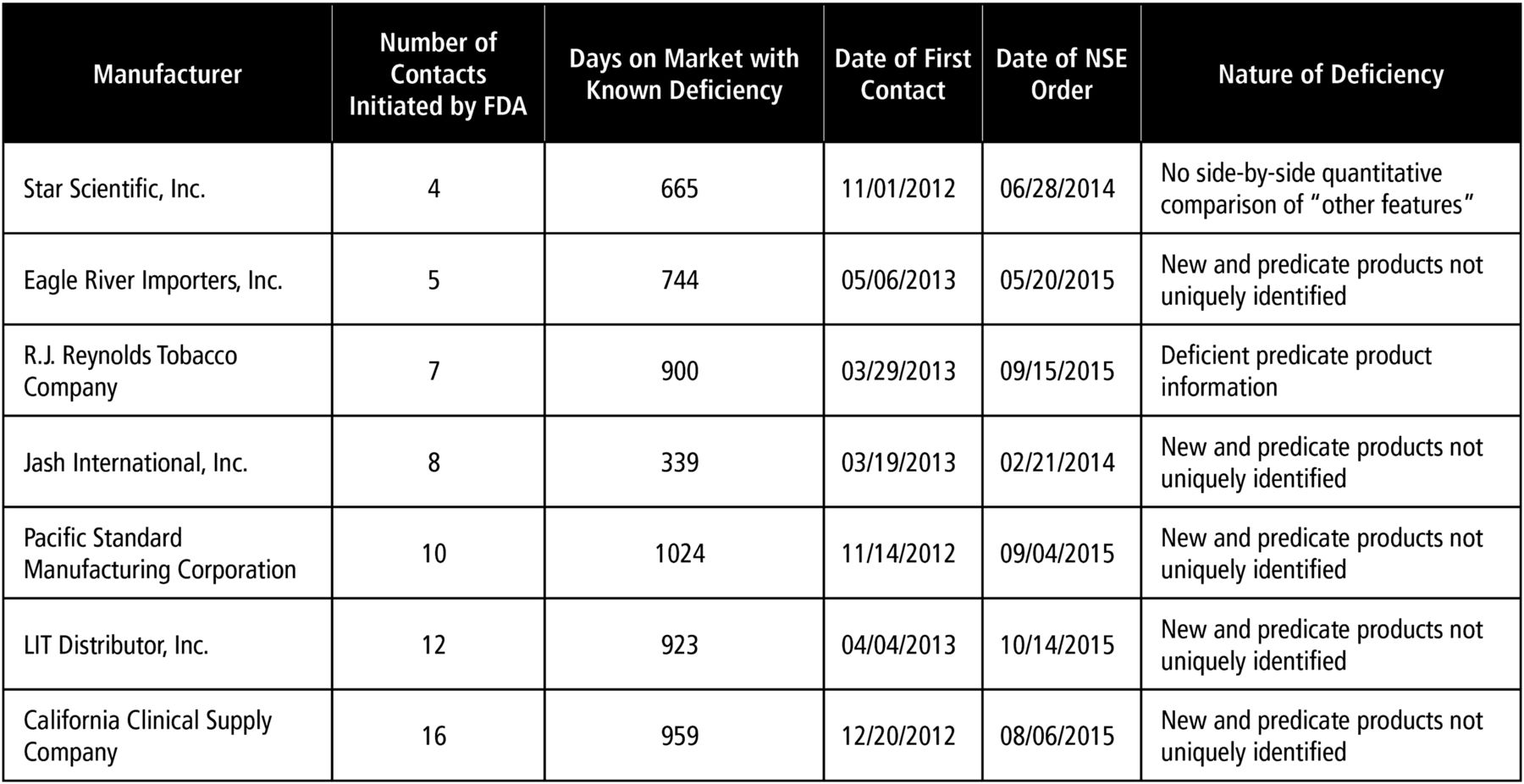
It is unclear how many other fatally deficient Provisional SE applications are pending before FDA. According to the agency, there have been significant deficiencies in nearly all the SE applications it has reviewed.12 ,13 ,15 ,21 ,29 The agency's Provisional NSE orders show the scope of these deficiencies (figure 4). For example, FDA orders rejecting 10 Provisional SE applications in May 2015 revealed that neither the new products nor the predicate products were uniquely identified despite at least five attempts to contact the manufacturer.30 In August and September 2015, NSE orders were issued for additional Provisional SE products that did not adequately identify the predicate or new products. The August NSE orders were issued to a manufacturer who did not respond to the agency's 16 attempted contacts over the course of 2 years.31 The September NSE orders were issued to a manufacturer who had notified the agency several years prior that the company had gone out of business.32 Even after receiving this notification, the agency continued to request either supplementary information or a formal withdrawal 10 times over a period of more than 2 years.32 In another case, rather than selecting an individual predicate product for its SE applications, as is required by the Tobacco Control Act, RJ Reynolds (RJR) submitted SE applications identifying ‘a composite of all cigarettes commercially marketed in the United States as of 15 February 2007’.33 Eventually, the agency instructed the manufacturer to amend its SE applications more than 2 years after they were submitted. When such predicate products were finally identified (after multiple requests from FDA), they clearly differed in meaningful ways from the new products. (One new product contained a crushable menthol capsule not found in the predicate product.) There is no reason it should have taken FDA years to act on these applications. RJR's decision to use the Provisional SE pathway was dubious, given the obvious weakness of its argument for SE. But because it took so long for FDA to act, this gambit enabled these products to stay on the market for more than 4 years.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
FDA response to Provisional SE report deficiencies. FDA, US Food and Drug Administration; SE, Substantial Equivalence.
In each of these cases, there was a significant delay between the submission of a Provisional SE application and the first contact between the agency and the applicant (figure 4). This delay is a direct result of the agency's prioritisation, not a result of a lack of agency resources. This is evidenced by the significantly shorter delay in contacting Regular SE applicants, and the fact that even though the Provisional SE applications were received first, Regular SE applicants were contacted 11 months earlier. The first Provisional SE applications were submitted to the agency in November 2010, but the first requests for additional information were sent to manufacturers in December 2012, a 25-month delay.17 For Regular SE applications, the first submissions were received in late March 2011, but the first requests for additional information were sent in January 2012, only a 10-month delay.23 The interconnectedness of the agency's priority of review, and its slow pace of action is important. Together, they have prevented the agency from making any appreciable progress in clearing its massive backlog of Provisional SE applications.
FDA's overly generous approach to clearly deficient applications further delays an already slow review process. More problematically, it establishes a culture in which there is seemingly no expectation that tobacco product manufacturers comply with the statutory requirements, or FDA's guidance. If an application is found to be deficient—even in fundamental ways—FDA will provide the manufacturer with repeated opportunities to modify the application. With the development of a new regulatory scheme, it is not unreasonable for the agency to provide initial feedback and request supplementary information on industry applications. However, it is not FDA's responsibility to do the industry's work. Because the agency is devoting significant resources to accommodating egregiously—and perhaps deliberately—deficient applications, it is more difficult for FDA to pursue other regulatory actions that might significantly impact public health.
Misplaced priority #3: FDA inaction is undermining the premarket review process by allowing the industry to market new, unauthorised products
Even though FDA's slow review of Provisional SE applications has allowed the tobacco industry to continue marketing products that were available before 22 March 2011, manufacturers have been eager to introduce new products after that date. Some companies have even introduced brand new products to the market, or significantly modified existing products, without any authorisation from FDA, in clear violation of the Tobacco Control Act. Yet, FDA has, thus far, failed to take any public enforcement action against these companies.
For example, Philip Morris, USA, launched Marlboro Black and Marlboro Black Menthol in December 2011.34 ,35 According to filings with the Securities and Exchange Commission, the company also began marketing Marlboro NXT in 27 states in September 2012, and expanded the marketing to the remaining 23 states in July 2013.36 ,37 The manufacturer subsequently launched Marlboro Edge in October 2013, and Marlboro Midnight in 2015.39–40 Similarly, RJR introduced a new brand of heat-not-burn cigarettes, called Revo, in February 2015, and also introduced two new conventional cigarettes, Camel White and Camel White Menthol, in April 2015.41–43 Public statements by the tobacco companies leave no doubt that these products are ‘new products’ that ought to be subject to premarket review. There is also no doubt that FDA has not issued orders authorising the marketing of these products (all such orders are made public by the agency). Yet there has been no indication that FDA has taken any enforcement action related to the marketing of these products, and most of them are still available in retail stores. RJR discontinued Revo because the product did not meet the company's expectations, and the manufacturer has also removed Camel White from the market without a public announcement, but the other products remain available.44 ,45
In addition to these widely publicised new product releases, smokeless tobacco product manufacturers are making significant modifications to their products, the types of modifications that must be cleared with FDA prior to marketing. For example, between 2010 and 2011, Marlboro Snus introduced a larger ‘round tin’, similar in appearance to conventional moist snuff.46 The snus pouches in the ‘round tin’ packaging are larger and contain a higher moisture content than the previous version.46 ,47 These changes appear to play a significant role in the level of nicotine and tobacco-specific N-nitrosamines in these products, and therefore have substantial implications for public health.47 Because FDA does not make such information available, it is not clear whether Altria (parent company of Philip Morris, USA) submitted Provisional SE applications for Marlboro Snus products by the 2011 deadline. If so, these SE applications would have been inappropriate, as these products were clearly modified after 15 February 2007, and any changes after that date that ‘raise different questions of public health’ require a PMTA application, not an SE application. As FDA has yet to act on any SE applications for Marlboro Snus products, it is therefore the case that either (1) Altria is abusing the SE process, aided by FDA's failure to review Provisional SE applications in a timely manner, or (2) Altria is making significant product modifications without the required FDA review, and FDA has failed to enforce the law in a timely way.
Although the agency ought to be able to, FDA is not identifying new products entering the market without marketing orders. One readily apparent solution to this problem is for the agency to implement its authority to require a track-and-trace programme for tobacco products. Such a system, where each individual tobacco product has a unique identification code or computer chip that allows the agency to track the product from the assembly line to a consumer's hands, is contemplated by the Tobacco Control Act, and public health groups have asked FDA to implement this authority.48 ,49 A thorough track-and-trace system would give FDA a robust regulatory tool to prevent unauthorised products from entering the market.
It is equally important for the agency to monitor significant changes to existing products, particularly when these changes result in increases to addictive and carcinogenic constituents. Requiring premarket review of new tobacco products is a basic pillar of the Tobacco Control Act's regulatory structure. If FDA permits companies—including the major cigarette manufacturers—to modify their products without completing the required premarket review process, why would any company comply with the law? The lack of quick and aggressive FDA action against companies that are brazenly introducing new products and modifying existing ones undermines the premarket review process and fails to protect public health.
Policy recommendations and conclusion
FDA has significant opportunities to improve the premarket review process to better protect public health. The authors suggest the following steps for immediate, impactful changes:
FDA should reverse its prioritisation of Regular over Provisional SE applications. Provisional SE products that are currently on the market have been given a free pass for more than 5 years, despite the fact that many of them likely do not meet the legal test for SE.
FDA should immediately identify Provisional SE applications that have failed to identify either the new or predicate product. Such applications that have not already been corrected should be given only one attempt to correct the deficiency before the agency issues an NSE order, creating a greater incentive for the industry to submit complete applications.
FDA should establish benchmarks for the length of time a deficient application may remain pending, and the number of times FDA will contact an applicant to seek additional information. When scientific information cannot be corrected within 90 days, or the applicant fails to respond to requests for additional information, the agency should promptly issue an NSE order rather than making additional attempts to solicit amendments. The agency must no longer tolerate gross deficiencies; the tobacco industry has had years to correct deficient pending applications, and there has been ample information released regarding the types of deficiencies that result in NSE orders. This is true even for applicants who have not been contacted regarding deficiencies. The statute requires FDA to act based on information provided by the applicant, and if that information is deficient, FDA is required to reject an application. Therefore, it is the applicant's responsibility, not FDA's, to ensure that an application is complete and accurate.
FDA should begin monitoring the tobacco product market more closely. In order to do so, the agency should implement a robust track-and-trace system. FDA should also begin regularly reviewing tobacco industry public announcements and biannual manufacturer registrations with the agency so that it can quickly identify and take enforcement action to remove any products that enter the market without authorisation.
To increase the participation of the public health community, FDA should make its premarket review activities more transparent, and provide educational materials designed for the public health community. For all NSE orders, FDA should publish the full applications with minimal redactions (only those required by law). FDA should also publish the product names from all Provisional SE applications that have been submitted. Similarly, FDA should identify any products that have been certified as grandfathered tobacco products. Providing this information will allow the public health community to supplement the agency's monitoring of the tobacco product marketplace and identify products entering the market without authorisation.
FDA's premarket review authority rests on the premise that without the express authorisation of the agency, no new tobacco product can enter the marketplace. Unless FDA truly controls entry to the tobacco product market, as the Tobacco Control Act clearly requires, the public health gains of a review process will be minimal. The agency has had nearly 7 years to implement a regulatory system, yet is has failed to meet this basic prerequisite for effective regulation. Instead, FDA has set priorities that undermine its ability to protect the public.
Rather than prioritising the review of Provisional SE products that have been allowed to remain on the market without any oversight, FDA has facilitated the introduction of additional tobacco products. Likewise, rather than guard the marketplace from illegal or otherwise unauthorised products, FDA has wasted resources by giving significant leeway to tobacco companies to correct applications that are grossly incomplete if not deliberately deficient. Significant reform is needed for FDA to satisfy its obligation to protect public health.
What this paper adds
While the Tobacco Control Act has ushered in an era of premarket review of new tobacco products, the US Food and Drug Administration's (FDA) implementation and enforcement of the premarket review requirements has not fulfilled the agency's obligation to protect public health.
FDA's decision to prioritise the review of Regular Substantial Equivalence (SE) applications over Provisional SE applications has allowed the industry to keep thousands of unreviewed provisional products on the retail market.
FDA is providing excessive opportunities to the tobacco industry to correct deficient submissions, leading to significant delays in removing unreviewed provisional products from the retail market.
Despite the premarket review requirements, there is evidence that the tobacco industry is introducing new products that have not been authorised by FDA, and the agency has yet to take an action to stop or prevent this practice despite the fact that the industry's actions have been made public.
Acknowledgments
The authors thank The Robert Wood Johnson Foundation for its recognition of the importance of legal expertise in the furtherance of public health policy. The authors also thank Dr Ruth Malone for her encouragement throughout the editing process.
Footnotes
Twitter Follow Desmond Jenson at @desmondjenson
Contributors DJ and JL conceived of this article. DJ wrote the first draft of the article. JL and MLB reviewed, edited and revised all drafts.
Funding This work was supported by The Robert Wood Johnson Foundation.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
↵i This FDA policy change and the tobacco industry's current legal challenge to the policy are not discussed in this paper. The public health implications of this important and potentially harmful policy warrant significant discussion by the public health community but, due to space limitations, this paper will not address this subject.