
Abstract
Gene signatures of drug-induced toxicity are of broad interest, but they are often identified from small-scale, single-time point experiments, and are therefore of limited applicability. To address this issue, we performed multivariate analysis of gene expression, cell-based assays, and histopathological data in the TG-GATEs (Toxicogenomics Project-Genomics Assisted Toxicity Evaluation system) database. Data mining highlights four genes—EGR1, ATF3, GDF15 and FGF21—that are induced 2 h after drug administration in human and rat primary hepatocytes poised to eventually undergo cytotoxicity-induced cell death. Modelling and simulation reveals that these early stress-response genes form a functional network with evolutionarily conserved structure and intrinsic dynamics. This is underlined by the fact that early induction of this network in vivo predicts drug-induced liver and kidney pathology with high accuracy. Our findings demonstrate the value of early gene-expression signatures in predicting and understanding compound-induced toxicity. The identified network can empower first-line tests that reduce animal use and costs of safety evaluation.
Similar content being viewed by others


Introduction
Drug toxicity and associated adverse events are critical issues in drug development: by a general estimate, 20–40% of novel drug candidates fail because of safety issues.1, 2, 3 Over the past 10 years, gene expression profiling has been introduced into drug development to predict and understand toxicity in pre-clinical settings, either as a stand-alone method4, 5, 6, 7 or integrated within systematic approaches.8, 9, 10, 11, 12, 13
In this work, we focus on the problem of identifying transcriptomics signatures that (a) predict toxicity prior to histopathological observations and (b) extrapolate between in vitro and in vivo settings and across species. Several studies have undertaken this task:14, 15, 16, 17 while making remarkable progress, they have illustrated two major challenges caused by limited data availability.18 First, most studies have derived signatures from small-scale experiments with one or few compounds. Such signatures suffer both from limited statistical power and from limited translatability to other compounds. At the same time, it is difficult to interpret ‘black-box’ signatures, which are statistically significant but not associated with biological function. Second, most studies have sought late-time signatures (≥24 h). Such signatures are likely to be downstream effectors of signalling networks, which are compound specific and highly dependent on the specific cellular context and the availability of complex regulatory elements, as recently revealed by the ENCODE project.19 In contrast, early-response genes (<24 h) may be less context specific and more generic, because many of them are essential transcription factors or regulators of stress response,20 and they are closer to the core machineries of ‘bow-tie’-like signalling networks.21
It is not feasible to identify early-response toxicity signatures from large-scale experiments with current bottom–up practices.22 Therefore, we took a top–down approach by analysing TG-GATEs (Toxicogenomics Project-Genomics Assisted Toxicity Evaluation system), a toxicogenomics database covering 170 compounds (Supplementary Table 1). The data comprise time-series gene expression, cell-based assay readouts and pathological records (Figure 1a). TG-GATEs is publicly available and is one of the most comprehensive data sets up to date.6, 23, 24, 25

Study design of TG-GATEs (Toxicogenomics Project-Genomics Assisted Toxicity Evaluation system) and the computational pipeline for data mining with TG-GATEs. (a) In TG-GATEs, 170 compounds are tested in up to three doses (including corresponding vehicle controls) in three systems: human primary hepatocytes, rat primary hepatocytes and rat in vivo. In in vitro experiments, compounds are administrated once at 0 h (red triangle). Gene expression profiling and Pico-Green DNA quantification assays are performed at three time points (2, 8 and 24 h). In in vivo experiments, compounds are dosed either once or repeatedly. Gene expression profiling and examination of pathology in liver and kidney take place at 3, 6, 9 and 24 h (single-dose) or at 4, 8, 15 and 29 days (repeated dose). Two biological replicates are available for most experiments. (b) The computational pipeline that integrates gene expression data, cell-assay readouts and pathology records to identify early signatures of in vitro and in vivo toxicity. Details are described in the Results section.
Here we report an ab initio analysis of TG-GATEs using a novel computational pipeline (Figure 1b). We start by reporting data that support the generality of early-response genes. Next, we describe how integrative analysis of differential gene expression and cellular assay data revealed a consensus set of cytotoxicity signatures in human and rat primary hepatocytes. We provide evidence that the signatures form an evolutionarily conserved functional network that is responsible for early stress response. Finally, we confirm the network’s predictive power for hepatic and renal pathology in a short-term (<30 days) study in vivo.
Materials and methods
Data pre-processing
Raw data were downloaded from the TG-GATEs website (http://toxico.nibio.go.jp). Gene expression was profiled with Affymetrix Human Genome U133 PLUS 2.0 (Santa Clara, CA, USA) or Rat Genome 230 2.0 chips (Santa Clara, CA, USA). For each in vitro experiment, a Pico-Green fluorescence assay was performed, which quantitatively measures the total DNA content of cells. For in vivo experiments, histopathology in liver and kidney was assessed by pathologists (Supplementary Table 2).
Expression data were pre-processed with the MAS5 method.26 Differential gene expression profiles were determined by linear models using limma,27 and expressed in logarithmic (base 2) fold changes (logFC). For in vitro samples, cytotoxicity was determined by the difference of total DNA content between compound-treated samples and the time-matched control samples. Large reduction of DNA content indicates strong cytotoxicity.
Identification of early-response gene signatures of cytotoxicity
We merged all differential gene expression profiles of human samples, irrespective of compound, dose and time, into one matrix. Unsupervised clustering was performed and classified the samples into three groups. The statistical association between the groups and DNA content was determined by one-way analysis of variance.
A cytotoxicity matrix was constructed for each compound by projecting the three sample groups (weak, moderate and strong, referring to corresponding cytotoxicities) onto a two-way matrix of dose and time. Gene signatures were derived from progressive profiles, where compound treatment causes weak, moderate and strong cytotoxicity at 2, 8 and 24 h, respectively. Hierarchical linear models28 were fitted to capture early-response signatures (Supplementary Methods).
We validated the signatures by analysing rat data with the same procedure, not involving any information from human data.
Modelling and simulation with Boolean networks
Inspired by a recently developed method, the Boolean Network Ensembles, which generates semiquantitative time-response simulations from the network structure,29 we developed an algorithm that generates time-response profiles of expected node occupancy fractions given initial states (Supplementary Methods). Intuitively, the occupancy fraction of a node is the expected probability that the node will be ON. The time evolution of occupancy fractions simulates the dynamics of the network.
Predicting liver and kidney pathology in vivo
In vivo expression and histopathological records were randomly split into a training set (80%) and a test set (20%). Radial-kernel support vector machines (SVMs) were trained by 10-fold cross-validation using LIBSVM.30 Optimal parameters of SVMs (C and gamma) were determined by grid search. Accuracy was measured by the F1 score, which combines precision and recall of predictions (Supplementary Methods).
Results
Early-response genes are more generic than late-time induced genes
We performed differential gene expression analysis to human gene expression profiles in TG-GATEs, by comparing compound-treated cells with the time-matched controls (Figure 1bA).
We investigated how global differential expression patterns change over time. In particular, we focused on the differentially expressed genes (DEGs) with |logFC|>0.5 and multiple-testing adjusted P<0.05 (Benjamini–Hochberg method). Notably, irrespective of compound dose, there are generally fewer genes induced at 2 h than at 24 h (Figure 2a).

Across the TG-GATEs (Toxicogenomics Project-Genomics Assisted Toxicity Evaluation system) data set, there are fewer early-response genes but they are more generic than late-response genes. (a) Number of differentially expressed genes (DEGs) induced by compounds in human primary hepatocytes, stratified by dose and time. Numbers of DEGs induced by TG-GATEs compounds are represented in boxplots, with each blue dot representing one compound. Black dots indicate the median number of DEGs. (b) The generality score of DEGs stratified by dose and time. The score measures how often a DEG is induced by different treatments. DEGs with higher scores are more generic (less specific) than DEGs with lower scores. The scores are normalized to the mean value of DEGs at 24 h. Bar heights and error bars indicate the average score and the standard deviation, respectively.
Given the number of DEGs and the number of DEG-inducing treatments at each time point, we asked whether DEGs are induced by similar numbers of treatments at three time points. To test this, we calculated the generality score for each DEG, representing how often it is induced by different treatments. DEGs with higher scores are more generic (less specific) than DEGs with lower scores, as they are induced by more treatments. A comparison of the scores shows that genes induced at 2 h are likely to be more generic than genes induced at 8 or 24 h (Figure 2b). A similar pattern was observed in the rat data (Supplementary Figure 1).
Although there are generally fewer genes induced at 2 h, these genes tend to be more generic in the sense that they are modulated by multiple treatments. This observation guided us to focus on early-response genes as potential generic toxicity signatures in subsequent analyses.
Early-response toxicity signatures identified in human hepatocytes
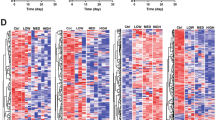
To identify early toxicity signatures in vitro, we set out to determine which combinations of compound, dose and time cause cytotoxicity. We took a data-driven approach to address this question by integrating gene expression data with the results of DNA quantification assays (Figure 1bB). Unsupervised clustering of differential expression profiles revealed that compound-treated samples are classified into three groups with distinct features (Figure 3a). Statistical analysis showed that the groups are significantly correlated with decreasing DNA content, or equivalently, with increasing cytotoxicities (Figure 3b).

Early-response signatures identified in human primary hepatocytes. (a) Unsupervised clustering of differential expression profiles revealed three groups (separated by black lines). Each row of the matrix is a differential expression profile induced by a certain combination of compound, dose and time. Columns are frequently induced genes, which were induced by >5% of compound treatments. (b) The three sample groups are significantly associated with decreasing DNA content and increasing cytotoxicities: samples in the weak group show no significant DNA loss (P=0.60, Student’s t-test). The moderate group is associated with slight DNA content loss (−1.28% compared with the weak group, P=8.0E–4, 95% confidence interval (CI; −2.11%,−0.46%), Tukey’s method), and the strong group showed the most substantial loss of DNA (−6.97% compared with the weak group, P<1E–7, 95% CI (−8.44%, −5.51%), Tukey’s method). The three asterisks (***) indicate that the P-value of comparing DNA content of two adjacent groups is smaller than 0.001. (c) Example toxicity matrices of vitamin A and of nitrofurantoin, with the progressive profile induced by high dose of nitrofurantoin being highlighted. Green, yellow and red cells indicate weak, moderate and strong toxicity levels respectively. Empty cells: no data available. (d) Dynamic patterns of early-response cytotoxicity signatures in human. Thin lines represent expression changes in each treatment, and thick lines represent the arithmetic mean. Error bars indicate standard errors of the mean.
We assigned one of the three cytotoxicity levels (weak, moderate or strong) to each sample, which is associated with a unique combination of compound, dose and time. Subsequently, we built a cytotoxicity matrix for each compound, presenting how its cytotoxicity varies with dose and time (Figure 1bC). Cytotoxicity matrices of all compounds tested in human primary hepatocytes are given in Supplementary Figure 2, and two of them are illustrated in Figure 3c as examples: vitamin A, which is nontoxic over the entire tested time and dose range, and nitrofurantoin, which is an antibiotic that showed increasing toxicity both along the dose gradient and with time.
To identify early cytotoxicity signatures, which are detectable before the toxicity is visible at the molecular or phenotypic level, we focused on profiles of the progressive type, like the one described for nitrofurantoin, with weak, moderate and strong cytotoxicities at 2, 8 and 24 h, respectively (Figure 1bD). We asked if there are genes that are robustly induced at 2 h in such profiles, arguing that they may be predictive signatures of the cytotoxicity outcome at 24 h (Figure 1bE).
Statistical analysis identified five genes that are significantly upregulated at 2 h in progressive profiles: EGR1, GDF15, ATF3, FGF21 and IL8; and one gene that is downregulated: TOB2. We note that these genes are induced by compounds of diverse chemical and pharmacological properties (Table 1). Nevertheless the genes show consistent temporal expression patterns in progressive profiles. In contrast, no significant patterns were detected when their expression profiles were examined in treatments causing no or weak toxicity up to 24 h (Supplementary Figure 3).
Conserved early cytotoxicity signatures between rat and human
The analysis procedure described above was then applied to data from rat primary hepatocytes. We emphasize again that no information obtained from the human data were carried over.
Compound-induced expression profiles were classified into four groups in rat (Supplementary Figure 4). Three of them resemble the groups in human (weak, moderate and strong): they are associated with distinct expression profiles, and are significantly correlated with increasing cytotoxicities (Figure 4a). The fourth group only contains eight samples treated by either acetaminophen (paracetamol) or phenobarbital, both non-genotoxic carcinogens (NGTX) inducing tumour formation in rodent models.31, 32 Samples in this group contrast strongly with other treatments as judged by their gene expression profiles (Supplementary Figure 4). They are associated with loss of total DNA, in line with previous observations.33

Four early-response signatures were validated in rat. (a) The four groups of rat samples are significantly associated with cytotoxicities measured by DNA content. NGTX: non-genotoxic carcinogens. The three asterisks (***) indicate that the P-value of comparing DNA content of two adjacent groups is smaller than 0.001. (b) Venn diagrams comparing compounds that induce progressive profiles in human and rat, and comparing frequently induced genes in the two species. (c) Temporal patterns of the four human orthologs that are also signatures in rat. See caption of Figure 3d for legends.
We built cytotoxicity matrices for all compounds that were tested in rat primary hepatocytes (Supplementary Figure 5), and focused again on the progressive profiles. Comparing compound sets inducing progressive profiles in human and rat, we found only two overlapping compounds (Table 1 and Figure 4b), no more than the random expectation (P=0.46 by bootstrapping). The substantial lack of overlap may reflect the distinct bioavailability and different modes of action of compounds in the two species. In contrast, the overlap of frequently induced genes (defined as DEGs that are induced in >5% of samples) is far stronger (81 genes; Figure 4b, P=9.0E–6 by bootstrapping). The two comparisons suggest that although human and rat hepatocytes exhibit distinct temporal responses to toxicity-inducing compounds, the underlying mechanism is conserved to some extent.
We applied statistical analysis to detect early-response genes from progressive profiles in rat. Notably, out of the six genes found in the human signatures, four orthologous genes were identified as early cytotoxicity signatures in rat: Egr1, Atf3, Gdf15 and Fgf21 (Figure 4c). Sequence analysis revealed high similarities between the orthologs (Supplementary Figure 6A). The two human genes that were not selected as signature genes in rat either do not have a rat ortholog (IL8, Supplementary Figure 6B) or just fail to achieve the predefined statistical significance (TOB2, Supplementary Figure 6C).
The observation that four early-response genes show consistent compound-induced activation in human and rat hepatocytes suggests that they are likely to be evolutionarily conserved, functionally linked and intrinsic to the cell’s stress-response system.
An early-response network with conserved structure and intrinsic dynamics
Compound-induced expression changes of EGR1, GDF15, FGF21 and ATF3 in vitro are summarized in Figure 5a. The side-by-side comparison reveals strikingly conserved dynamics in human and rat: the induction of all four genes at 2 h, followed by decay of EGR1 and persistent activation of FGF21, GDF15 and ATF3.

Dynamics of the early-response network is encoded by its structure. (a) Dynamics of the network in human (left) and rat (right), showing their arithmetic mean (lines) and standard errors of the mean (error bars). (b) The Boolean network model. Four genes are connected with thick lines embodying binary interactions. Solid-arrow tip: positive trans-activation; solid-circle tip: indirect activation; bar: transcriptional inhibition. Biological functions are given in rectangular nodes, with genes connected to them with thin lines. Solid-circle tips indicate positive effects, and bars indicate negative effects. Only gene nodes and their interactions are used for modelling and simulation. (c) Simulated dynamics using the Boolean network model with the initial state IS1 (top) and IS2 (bottom). Random noise is added to facilitate visualization.
Literature search34, 35, 36, 37, 38, 39, 40, 41, 42 allowed us to construct a functional network of the four genes (Figure 5b). The well-connected network is small but non-trivial: it has an auto-inhibition loop (EGR1), an auto-activation loop (ATF3), a negative feedback loop (EGR1 and ATF3), and a feed-forward loop (EGR1, ATF3 and GDF15). Such components, known as network motifs, can encode dynamic behaviour of networks.43
We hypothesized that the conserved dynamics is intrinsic to the network, and tested this with a Boolean network model (Figure 1bG).44, 45 Boolean networks, as the name suggests, are defined as a set of nodes and edges. All nodes (representing, for example, genes, proteins or small molecules) are in either one of the two states: ON and OFF, and edges (interactions) between nodes are characterized as either activation or inhibition. Update functions change the states of nodes as a function of their incoming activating and inhibiting edges. Computational analysis of a Boolean network produces all the possible steady states of the network, namely the states of the network for which no further updates of node states are taking place, that is, when the network is in equilibrium. In addition, the Boolean network formalism is in principle able to simulate the dynamics of a network given the initial states of all network nodes.
Given the network structure in Figure 5b, we identified two steady states (S):S1 (EGR OFF, ATF3, GDF15, and FGF21 ON) represents inactive (not induced) EGR1 and active (induced) ATF3, GDF15, and FGF21; S2 (all four genes OFF) represents all genes at the baseline level. Note that S1 matches the state of progressive profiles at 24 h (Figures 5a and b), and S2 matches the state of non-toxic treatments (Supplementary Figures 3A and B). The Boolean network model thus is in agreement with the two observed steady states.
Next, we simulated the dynamics of the network with two different initial states (IS): IS1 assumes all four nodes are initially ON, whereas IS2 assumes only EGR1 is ON and all the others are OFF (Figure 5c). The two alternatives were chosen because neither of them could be ruled out based on prior knowledge. Independent of the choice of initial states, simulation results match very well with observed dynamics: EGR1 induction gradually decays and is finally turned OFF. The other three genes either stay ON (IS1) or are induced from OFF to ON (IS2). This implies the network dynamics is encoded by its structure, and the Boolean network is a useful model to study this network in silico.
Subsequently, we tested the robustness of the network by deleting one edge a time and simulating the network dynamics with permutated networks. By iterating all single-edge mutations, we found that the deletion of any edge involving ATF3 (including the self-loop) dramatically alters the steady state and/or the dynamics of the network (Supplementary Figure 7). This implies that ATF3 is an essential gene of the network, and underscores the importance of ATF3 in the context of cellular response to compound-induced toxicity.42, 46
Early expression changes of the network demonstrate predictive power for pathological outcomes in vivo
Finally, we evaluated the predictive power of the early-response network for pathological outcomes in vivo (Figure 1bH). To this end, we trained SVM, an established tool for binary prediction.47
An SVM is constructed in two steps. First, it is trained with samples with binary labels (the training set). The SVM learns patterns from the data by finding the boundary (known as the hyperplane) in data space that best separates samples of two classes. Next, the SVM is used to predict labels for a new data set (the test set) and its accuracy is measured by comparing the predictions with true labels.
SVMs used in this study take treatment-induced differential expression of Egr1, Atf3, Gdf15 and Fgf21 at 3 h (the earliest time point in vivo) as input, and predict histopathological outcomes at all tested time points of the in vivo study. For each time point, one SVM was trained and tested for liver and kidney, respectively. We found that SVMs are able to predict pathological outcomes with high accuracies between 80 and 97% both in liver (Figure 6a) and in kidney (Figure 6b). This finding suggests that expression changes of the four early-response genes contain information that can predict short-term in vivo pathology.

Performance (F1-score) of support vector machines (SVMs) with four genes (network) as input and of SVMs with individual genes as input, in liver (a) and kidney (b).
To test whether the network’s predictive power is superior to that of single genes, we compared SVMs powered by the four-gene network against SVMs that use single genes as input (Figures 6a and b). The network-based SVM substantially outperformed single-gene competitors, confirming synergy between the genes.
Discussion
Large-scale databases such as the Drug Activity database of NCI-60 cell lines,48 Connectivity Map49 and ToxCast50 have been extensively explored to study compound-induced gene expression. However, few databases collect time series data, which is essential to identify early-response genes. We argue that such early-response genes can be valuable to understand and predict in vitro and in vivo toxicity. Our study presents a piece of evidence: although the four-gene network discussed here was identified in primary hepatocytes, it predicted liver and kidney pathology with good performance. Thus, by multivariate analysis of time-series data, it may be possible to overcome one of the biggest hurdles for predictive in vitro toxicity, namely the lack of conserved markers between in vitro and in vivo and across species. However, we emphasize that the marker gene set identified here, whereas translatable from cellular systems to rat in vivo, allows only for a broad categorization of potential risk. This should guide researchers in prioritizing candidates out of compound series, including more specific measurements to identify the mode of action.
Many parameters have been proposed as in vitro toxicity and tissue injury markers. Gerets et al.51 identified a battery of six cytotoxicity assays to screen pharmaceutical compounds in HepG2 cells using eight drugs. Recently, Bailey et al.52 evaluated 34 acute rat toxicity studies and proposed three novel candidate genes (GSTA, ARG1 and HPD) in addition to the established ALT as drug-induced liver injury biomarkers in rats. Complementary to these studies, we present here with the network of EGR1, ATF3, GDF15 and FGF21 a signature set detectable as early as 2 h after compound administration. We propose the network should be monitored in combination with established parameters for better toxicity prediction.
Biological functions and biomarker potentials of EGR1, ATF3, GDF15 and FGF21 have been sporadically proposed for compound-induced in vitro and in vivo toxicity. EGR1, arguably the best-studied gene among them, is stimulated by many extracellular molecules. It links signalling cascades controlling cellular proliferation and apoptosis.53 Bioinformatics and mRNA expression analyses showed Egr1 mRNA activation is dependent on the activation of the Ras-Raf-Mek-Erk signalling pathway.54, 55 Transforming growth factor-β, an important mediator of the cellular response to external stimuli and xenobiotics, activates EGR1 via SMAD3.56 Upregulation of EGR1 was described as an adaptive measure to attenuate sulindac sulphide-mediated cytotoxicity in human intestinal epithelial cells.57 In vivo studies revealed that EGR1 is essential for ethanol-induced58 or cholestatic59 liver injury. Our study provides further evidences that early activation of EGR1, presumably an adaptive reaction against cell death, is a signature of compound-induced toxicity. Its expression alone, however, is not sufficient for prediction unless the states of the other genes in the network are known.
ATF3, a direct transcriptional target of EGR1, acts as a central hub coordinating cellular stress pathways.46, 60 It has a key role in the transforming growth factor-β-SMAD pathway, providing a convergence point for the joint control of epithelial cells by multiple stress response pathways.61 Therefore, it has been proposed as marker for a variety of stressed tissues including liver, heart, brain62 and nerve.63 Our findings underpin its role in stress response and highlight its synergy with other genes of the network.
The other two members of the network both protect cells from apoptosis. GDF15, a transforming growth factor-β superfamily member, protects heart from ischemia/reperfusion injury.64 Its rapid induction was shown in various models of liver, lung and kidney injury.65, 66 Serum levels of FGF21, a secreted protein, were recently proposed as a biomarker for liver and kidney diseases.67, 68
Although their functions have been individually characterized, the hitherto undescribed coherent dynamics of the four genes induced by distinct compounds in human and rat suggest that they are more closely linked than previously thought. The network may act both as a convergence point of upstream stress-sensing pathways and as a core module coordinating downstream responses. Boolean network modelling supports the notion that the network’s intrinsic dynamics is encoded by its structure. It presents a hypothesis to explain the conserved dynamics and the generality across compounds, organ types, and even species. We believe this hypothesis should be challenged by studies with other cell types, and studies in model organisms beyond rat and human.
In conclusion, we report EGR1, ATF3, GDF15 and FGF21 as a consensus early signature of in vitro and in vivo toxicity in human and rat. It was essential to focus on early time points, because at this stage there seems to be high conservation of general stress-response signals, which diverge in later time points. Our findings demonstrate the translational value of multivariate time-series data in toxicity studies and the potential of early-response genes as predictive toxicity signatures. We recommend monitoring the network in first-line compound screenings to increase the efficiency of safety evaluations and to reduce costs and animal use.
References
Kola I, Landis J . Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 2004; 3: 711–716.
Arrowsmith J . Trial watch: phase II failures: 2008—2010. Nat Rev Drug Discov 2011a; 10: 328–329.
Arrowsmith J . Trial watch: phase III and submission failures: 2007—2010. Nat Rev Drug Discov 2011b; 10: 87.
Waring JF, Ciurlionis R, Jolly RA, Heindel M, Ulrich RG . Microarray analysis of hepatotoxins in vitro reveals a correlation between gene expression profiles and mechanisms of toxicity. Toxicol Lett 2001; 120: 359–368.
Waters M, Stasiewicz S, Alex Merrick B, Tomer K, Bushel P, Paules R et al. CEBS—Chemical Effects in Biological Systems: a public data repository integrating study design and toxicity data with microarray and proteomics data. Nucleic Acids Res 2008; 36 (Suppl 1): D892–D900.
Kiyosawa N, Ando Y, Watanabe K, Niino N, Manabe S, Yamoto T . Scoring multiple toxicological endpoints using a toxicogenomic database. Toxicol Lett 2009; 188: 91–97.
Yuan B, Wang J, Cao H, Sun R, Wang Y . High-throughput analysis of the mutagenic and cytotoxic properties of DNA lesions by next-generation sequencing. Nucleic Acids Res 2011; 39: 5945–5954.
Steiner G, Suter L, Boess F, Gasser R, Cristina de Vera M, Albertini S et al. Discriminating different classes of toxicants by transcript profiling. Environ Health Perspect 2004; 112: 1236–1248.
Heijne WH, Kienhuis AS, van Ommen B, Stierum RH, Groten JP . Systems toxicology: applications of toxicogenomics, transcriptomics, proteomics and metabolomics in toxicology. Expert Rev Proteomics 2005; 2: 767–780.
Butcher EC . Can cell systems biology rescue drug discovery? Nat Rev Drug Discov 2005; 4: 461–467.
Chiang A, Butte A . Data-driven methods to discover molecular determinants of serious adverse drug events. Clin Pharmacol Ther 2009; 85: 259–268.
Cami A, Arnold A, Manzi S, Reis B . Predicting adverse drug events using pharmacological network models. Sci Transl Med 2011; 3: 114ra27.
Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012; 486: 361–367.
Fielden MR, Eynon BP, Natsoulis G, Jarnagin K, Banas D, Kolaja KL . A gene expression signature that predicts the future onset of drug-induced renal tubular toxicity. Toxicol Pathol 2005; 33: 675–683.
Powell CL, Kosyk O, Ross PK, Schoonhoven R, Boysen G, Swenberg JA et al. Phenotypic anchoring of acetaminophen-induced oxidative stress with gene expression profiles in rat liver. Toxicol Sci 2006; 93: 213–222.
Martin MT, Brennan RJ, Hu W, Ayanoglu E, Lau C, Ren H et al. Toxicogenomic study of triazole fungicides and perfluoroalkyl acids in rat livers predicts toxicity and categorizes chemicals based on mechanisms of toxicity. Toxicol Sci 2007; 97: 595–613.
Nioi P, Pardo IDR, Sherratt PJ, Snyder RD . Prediction of non-genotoxic carcinogenesis in rats using changes in gene expression following acute dosing. Chem Biol Interact 2008; 172: 206–215.
Kramer JA, Sagartz JE, Morris DL . The application of discovery toxicology and pathology towards the design of safer pharmaceutical lead candidates. Nat Rev Drug Discov 2007; 6: 636–649.
Consortium TEP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74.
Activation of Immediate Early Genes By Drugs of Abuse. In: Grzanna R, Brown R eds. NIDA Research Monograph Series. US National Institute on Drug Abuse, 1993.
Citri A, Yarden Y . EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 2006; 7: 505–516.
Van Hummelen P, Sasaki J . State-of-the-art genomics approaches in toxicology. Mutat Res-Rev Mutat 2010; 705: 165–171.
Kiosawa N, Shiwaku K, Hirode M, Omura K, Uehara T, Shimizu T et al. Utilization of a one-dimensional score for surveying chemical-induced changes in expression levels of multiple biomarker gene sets using a large-scale toxicogenomics database. J Toxicol Sci 2006; 31: 433–448.
Hirode M, Omura K, Kiyosawa N, Uehara T, Shimuzu T, Ono A et al. Gene expression profiling in rat liver treated with various hepatotoxic-compounds inducing coagulopathy. J Toxicol Sci 2009; 34: 281–293.
Uehara T, Minowa Y, Morikawa Y, Kondo C, Maruyama T, Kato I et al. Prediction model of potential hepatocarcinogenicity of rat hepatocarcinogens using a large-scale toxicogenomics database. Toxicol Appl Pharmacol 2011; 255: 297–306.
Gautier L, Cope L, Bolstad B, Irizarry RA . affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004; 20: 307–315.
Smyth G . Limma: Linear Models for Microarray Data. In: Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S (eds) Bioinformatics and computational biology solutions using R and Bioconductor. Springer: New York, 2005.
Gelman A, Hill JH . Data Analysis Using Regression and Multilevel/Hierarchical Models. Cambridge University Press: New York, 2007.
Jack J, Wambaugh JF, Shah I . Simulating quantitative cellular responses using asynchronous threshold Boolean network ensembles. BMC Syst Biol 2011; 5: 109.
Chang C-C, Lin C-J . LIBSVM: a library for support vector machines. ACM TIST 2011; 2: 1.
Holsapple MP, Pitot HC, Cohen SH, Boobis AR, Klaunig JE, Pastoor T et al. Mode of action in relevance of rodent liver tumors to human cancer risk. Toxicol Sci 2006; 89: 51–56.
Hasegawa R, Furukawa F, Toyoda K, Jang JJ, Yamashita K, Sato S et al. Study for tumor-initiating effect of acetaminophen in two-stage liver carcinogenesis of male F344 rats. Carcinogenesis 1988; 9: 755–759.
Deutsch WA, Kukreja A, Shane B, Hegde V . Phenobarbital, oxazepam and Wyeth 14,643 cause DNA damage as measured by the Comet assay. Mutagenesis 2001; 16: 439–442.
Baek SJ, Kim J-S, Nixon JB, DiAugustine RP, Eling TE . Expression of NAG-1, a transforming growth factor-beta superfamily member, by Troglitazone requires the early growth response gene EGR-1. J Biol Chem 2004; 279: 6883–6892.
Bottone FG, Moon Y, Alston-Mills B, Eling TE . Transcriptional regulation of activating transcription factor 3 involves the early growth response-1 gene. J Pharmacol Exp Ther 2005; 315: 668–677.
Kumbrink J, Gerlinger M, Johnson JP . Egr-1 induces the expression of its corepressor Nab2 by activation of the Nab2 promoter thereby establishing a negative feedback loop. J Biol Chem 2005; 280: 42785–42793.
Fisher J, Henzinger TA . Executable cell biology. Nat Biotech 2007; 25: 1239–1249.
Johnson CL, Weston JY, Chadi SA, Fazio EN, Huff MW, Kharitonenkov A et al. Fibroblast growth factor 21 reduces the severity of cerulein-induced pancreatitis in mice. Gastroenterology 2009; 137: 1795–1804.
Lee S-H, Krisanapun C, Baek SJ . NSAID-activated gene-1 as a molecular target for capsaicin-induced apoptosis through a novel molecular mechanism involving GSK3beta, C/EBPbeta and ATF3. Carcinogenesis 2010; 31: 719–728.
Yin X, Wolford CC, Chang Y-S, McConoughey SJ, Ramsey SA, Aderem A et al. ATF3, an adaptive-response gene, enhances TGF-beta signaling and cancer-initiating cell features in breast cancer cells. J Cell Sci 2010; 123: 3558–3565.
Loeffler I, Hopfer U, Koczan D, Wolf G . Type VIII collagen modulates TGF-beta-induced proliferation of mesangial cells. J Am Soc Nephrol 2011; 22: 649–663.
Giraldo A, Barrett OPT, Tindall MJ, Fuller SJ, Amirak E, Bhattacharya BS et al. Feedback regulation by Atf3 in the endothelin-1-responsive transcriptome of cardiomyocytes: Egr1 is a principal Atf3 target. Biochem J 2012; 444 (Pt 2): 343.
Alon U . An Introduction to Systems Biology: Design Principles of Biological Circuits. Chapman and Hall/CRC: Boca Raton, FL, USA, 2006.
Thomas R, Thieffry D, Kaufman M . Dynamical behaviour of biological regulatory networks—I. Biological role of feedback loops and practical use of the concept of the loop-characteristic state. Bull Math Biol 1995; 57: 247–276.
Kauffman S . Gene regulation networks: a theory for their global structure and behaviors. Curr Top Dev Biol 1971; 6: 145–182.
Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U . ATF3 and stress responses. Gene Expr 1999; 7: 321–336.
Cortes C, Vapnik V . Support-vector networks. Machine Learning 1995; 20: 273–297.
Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet 2000; 24: 236–244.
Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Sci Signalling 2006; 313: 1929.
Dix DJ, Houck KA, Martin MT, Richard AM, Setzer RW, Kavlock RJ . The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci 2007; 95: 5–12.
Gerets HHJ, Hanon E, Cornet M, Dhalluin S, Depelchin O, Canning M et al. Selection of cytotoxicity markers for the screening of new chemical entities in a pharmaceutical context: a preliminary study using a multiplexing approach. Toxicol In Vitro 2009; 23: 319–332.
Bailey WJ, Holder D, Patel H, Devlin P, Gonzalez RJ, Hamilton V et al. A performance evaluation of three drug-induced liver injury biomarkers in the rat: alpha-glutathione S-transferase, arginase 1, and 4-hydroxyphenyl-pyruvate dioxygenase. Toxicol Sci 2012; 130: 229–244.
Thiel G, Cibelli G . Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol 2002; 193: 287–292.
Sakamoto KM, Bardeleben C, Yates KE, Raines MA, Golde DW, Gasson JC . 5′ Upstream sequence and genomic structure of the human primary response gene, EGR-1/TIS8. Oncogene 1991; 6: 867–871.
Gineitis D, Treisman R . Differential usage of signal transduction pathways defines two types of serum response factor target gene. J Biol Chem 2001; 276: 24531–24539.
Chen S-J, Ning H, Ishida W, Sodin-Semrl S, Takagawa S, Mori Y et al. The early-immediate gene EGR-1 is induced by transforming growth factor-beta and mediates stimulation of collagen gene expression. J Biol Chem 2006; 281: 21183–21197.
Moon Y, Yang H, Kim YB . Up-regulation of early growth response gene 1 (EGR-1) via ERK1/2 signals attenuates sulindac sulfide-mediated cytotoxicity in the human intestinal epithelial cells. Toxicol Appl Pharmacol 2007; 223: 155–163.
McMullen MR, Pritchard MT, Wang Q, Millward CA, Croniger CM, Nagy LE . Early growth response-1 transcription factor is essential for ethanol-induced fatty liver injury in Mice. Gastroenterology 2005; 128: 2066–2076.
Kim ND, Moon J-O, Slitt AL, Copple BL . Early growth response factor-1 is critical for cholestatic liver injury. Toxicol Sci 2006; 90: 586–595.
Tanaka Y, Nakamura A, Morioka MS, Inoue S, Tamamori-Adachi M, Yamada K et al. Systems analysis of ATF3 in stress response and cancer reveals opposing effects on pro-apoptotic genes in p53 pathway. PLoS One 2011; 6: e26848.
Kang Y, Chen C-R, Massagué J . A self-enabling TGF-beta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 2003; 11: 915–926.
Chen B, Wolfgang CD, Hai T . Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by Gadd153/Chop10. Mol Cell Biol 1996; 16: 1157–1168.
Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K et al. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol Cell Neurosci 2000; 15: 170–182.
Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers Jr et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res 2006; 98: 351–360.
Hsiao EC, Koniaris LG, Zimmers-Koniaris T, Sebald SM, Huynh TV, Lee SJ . Characterization of growth-differentiation factor 15, a transforming growth factor beta superfamily member induced following liver injury. Mol Cell Biol 2000; 20: 3742–3751.
Zimmers TA, Jin X, Hsiao EC, McGrath SA, Esquela AF, Koniaris LG . Growth differentiation factor-15/macrophage inhibitory cytokine-1 induction after kidney and lung injury. Shock 2005; 23: 543–548.
Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 2010; 139: 456–463.
Lin Z, Zhou Z, Liu Y, Gong Q, Yan X, Xiao J et al. Circulating FGF21 levels are progressively increased from the early to end stages of chronic kidney diseases and are associated with renal function in Chinese. PLoS One 2011; 6: e18398.
Acknowledgements
We thank Marco Berrera for his help on sequence analysis. We thank Laurent Essioux, Tobias Heckel, Jean-Christophe Hoflack, Thomas Singer, and members of the bioinformatics team for support and discussions. We want to especially thank the TG-GATEs consortium for establishing the database and for making it publicly available. This work was supported by F Hoffmann-La Roche AG.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
JDZ and ME designed research; JDZ, NB and ME performed research; JDZ, NB and ABR contributed new reagents and analytic tools; JDZ and NB analysed the data; JDZ, NB, ABR and ME wrote the paper.
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zhang, J., Berntenis, N., Roth, A. et al. Data mining reveals a network of early-response genes as a consensus signature of drug-induced in vitro and in vivo toxicity. Pharmacogenomics J 14, 208–216 (2014). https://doi.org/10.1038/tpj.2013.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2013.39
Keywords
This article is cited by
-
Harnessing male germline epigenomics for the genetic improvement in cattle
Journal of Animal Science and Biotechnology (2023)
-
Gadolinium labelled nanoliposomes as the platform for MRI theranostics: in vitro safety study in liver cells and macrophages
Scientific Reports (2020)
-
Opportunities and challenges in phenotypic drug discovery: an industry perspective
Nature Reviews Drug Discovery (2017)
-
Patient-specific hepatocyte-like cells derived from induced pluripotent stem cells model pazopanib-mediated hepatotoxicity
Scientific Reports (2017)
-
Model-based contextualization of in vitro toxicity data quantitatively predicts in vivo drug response in patients
Archives of Toxicology (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}